这里是简单的一些介绍。详细的内容建议看官方手册。官方的文档既全面,又会更新,是第一手资料。这里是SAM文件格式以及SAMtools的一些简单的介绍,更详细内容移步SAMtools相关官方文档。
SAM文件格式
通常得到个体/供体(individual/donor)的测序序列(reads)时,由于测序技术的原因,得到的是打碎的小片段,并不知道每个read在参考序列上位置,所以要mapping到参考基因组上去,找到其在参考基因组上的位置。比对结果通常以文本形式按SAM(Sequence Alignment/Map)格式进行保存,为了便于存储和传输,常被压缩为BAM和CRAM二进制文件。SAM格式文件包含测序片段的所有比对信息。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
| @HD VN:1.3 SO:coordinate
@SQ SN:11 LN:134452384
@PG ID:bwa PN:bwa VN:0.7.12-r1039 CL:bwa sampe -a 1500 /share/home/gaojingyang/NGS_Data/Reference/ncbi36/11.fa 10_11_1.sai 10_11_2.sai 10_11_1.fq 10_11_2.fq
11-1911076 99 11 52531 29 6S93M1S = 52706 275 CTACATTTATTTTGTAGAAGTTCTTTCACCTTTATCAGAAGGTACTTTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTC C=CFFEFDHHH+HH;AGIJBJIHJHHIJJJJJJIIIJIJJFDJJIJJJFJ)DGGJJJGHIDDGAIBHGIEDAGC6AA2BDFBFCDE@DCDBD=DE(;DD0 XT:A:M NM:i:0 SM:i:29 AM:i:29 XM:i:0 XO:i:0 XG:i:0 MD:Z:93
11-2807908 163 11 52535 60 100M = 52733 298 TTTGTAGATGTTCTTTCACCTTTATCAGAAGGTACTTTCACCTTGATTCAACAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTATAATTCACCC C@@4=FDF#H>HHJEJIIIIJJJIFAIIJHJII?IJ>JC<GEIJJJIGHB-DJJJCJGJJJIEHJ)DJJF5DJGAHEGCB:CD-BDAAF,:EDDDDD(DD XT:A:U NM:i:4 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:4 XO:i:0 XG:i:0 MD:Z:8A42T37G7T2
11-3392818 99 11 52557 60 100M = 52668 211 TATCAGAAGGTACTTTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGAGATT =@CAF)ADHH>HHFJJHGH+GIC,JBGIGIJHJIIGIII;GI*JJIJF)DFEJHEIIHDJD?E@HJECF=D.HD(D<F;?<DDHHEDDD2DD@,FE)CBD XT:A:U NM:i:1 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:1 XO:i:0 XG:i:0 MD:Z:96T3
11-11241866 163 11 52562 60 100M = 52753 291 GAAGGTACATTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCAGT CCC4=FFF#?HHGHCJ+,<JJFI::JHIIJGFHEGIJJBJJJIEDJBEJD,IHIJIEEBJBHCIJGDIDDHDDDFECJ+BB58EDHC+BDFDA?A:DD(B XT:A:U NM:i:2 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:2 XO:i:0 XG:i:0 MD:Z:8T89C1
11-589542 99 11 52563 60 100M = 52699 236 AAGGTACTTTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTCGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCACTA C@BFFFFFAHHHHJJJIJJIJJ3J13JAJICIJIGIC(HFJJ)HJJ?EIJJHIJDIJ)JHH)CFGFBIFG=DF>,BH>EDHD;DCCC?C?D5@5DDDDDB XT:A:U NM:i:1 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:1 XO:i:0 XG:i:0 MD:Z:57T42
11-11179046 99 11 52566 60 100M = 52764 298 GTACTTTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCACTACAT 1C@DFDDFHHF<HE=IJC@JIJHJGHJHHHIH)JIJ:JJHJ9IGDIJ)JJIFI=;JGIEJ@JD@H5(HIHBFHG99CI>BHEDDGDDDDECDDDDDDDBD XT:A:U NM:i:0 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:100
11-12910752 163 11 52568 60 100M = 52761 293 ACTTTCACCTTGTTTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCACTACATGA CCCF1BDEHFHF+J<J,IGIJGJHIJH:)IJICJHC/JII9GJ9=J/?HFHCIHI@HACHIBCJ;H=>FHHF>EFBDFD>@ACFD<,BBAD,C<EDD5DC XT:A:U NM:i:1 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:1 XO:i:0 XG:i:0 MD:Z:12A87
11-2579510 163 11 52568 60 100M = 52763 295 ACTTTCACCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCACTACATGA @+BFFDFFHHF+AIJJEHJJCJHJIGH#@IJHIH?DIJJIHFIIJIF>0CIBJDJHIFHJ=GCJ(D8GC8CHDJDCDFDD2CF+DEDBB>:CDCADDBC; XT:A:U NM:i:0 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:100
11-10363294 99 11 52575 60 100M = 52830 354 CCTTGATTCAATAATAAGTTTGCATATTACAACCTTGTTGAATGTTGGTGTTATTCATCCATTCGTATTGCTATATGATATTCCACTACATGAATATGTC ?B@DFFFFHHFFD(JGHIJJCFJIFJJJIJJIHFJGJHJDGDHIDGJIJIG)JBG=JEIFDH?IJ=AGF>I(CCDCEDBD;DDHE@:DDEBFDDDCD0=A XT:A:U NM:i:1 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:1 XO:i:0 XG:i:0 MD:Z:51A48
11-11225740 99 11 52599 60 100M = 52722 222 TATTACAACCTTGTTGAATGTTGGTGTAATTCATCCATTCGTATTGCTATATGATATTCCACTACATGAATATGTCGGACTTCATTCCTCAGATCTATTG B1<DDDFFH+HDFIHGJJJJJIJJJJIG33GI:HJGJIJGIGIJJJ?JJJJIHJIDI7HI=JE@DDF@JDEDH@BE>CD6CCDE>C>ECDDDD>D>DBCD XT:A:U NM:i:0 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:100
11-2240062 99 11 52630 60 100M = 52730 200 CATCCATTCGTATTGCTATATGATATTCCACTACATGAATATGTCGGACTTCATTCCTCAGATCTATTGTTGATGAACACTTGAAATTTTTCCAGTTTTT @C?FAFFFHCH8HJJHIIHIIHJHFJJHIFJJ*;JAHJIJJBJJJIEF?IJI7I)DFJI&5HJ7HIA@EH<DJ;CEED>HC(DCDDD5CC5C:(DDDD2C XT:A:U NM:i:0 SM:i:37 AM:i:37 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:100
11-5366918 163 11 52654 60 100M = 52840 286 ATTCCACTACATGAATATGTCGGATTTCATTCCTCAGATCTATTGTTGATGAACACTTGAAATTTTTCCAGTTTTTAACCATTACAAACAATGCTGCTAT CBCDADDF+DFFCJIJI=G3JI:G#IJEJIGHEJJG@:?@DGGGGJA
|
SAM格式文件包含两部分,头部注释和比对信息部分。头部注释包含了该文件的基本信息,每行以“@”开始,主要有SAM格式的版本、所用参考基因组的名称与长度、测序样本的描述信息、所用比对软件的信息及相关数据处理的命令行等。
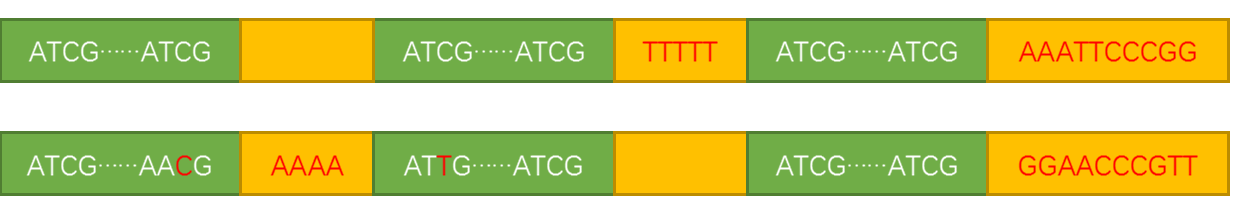
比对信息部分每行对应一个测序片段的比对信息,包含11个固定字段和可选字段。11个固定字段按顺序分别为,测序片段ID、位标识、所映射的染色体、映射的位置、映射的质量、CIGAR(比对情况)、配对片段的映射的染色体、配对片段映射的位置、模板长度、片段序列、序列质量信息以及一些可选字段。位标识的每个二进制位代表一个标识信息,主要是测序片段是否映射成功、映射方向等信息。CIGAR以字符串形式描述序列片段的具体比对信息,由数字+一个字母形成的串连接而成,数字为序列的长度,字母为这段序列与参考序列的比对形式。常用的比对形式有:
- “M”:匹配,该段序列与参考序列的碱基相对位,但可能包括错配。
- “D”:缺失,测序片段中相对于参考序列缺失此段碱基。
- “I”:插入,测序片段中相对于参考序列插入此段碱基。
- “S”:软剪切,测序片段比对得分为负,舍弃。
如30M5I25M5D25M10S意味着100bp的片段依次以30个碱基匹配、5个碱基插入、25个碱基匹配、5个碱基缺失,25个碱基匹配,10个碱基未匹配,具体可参照下图。
每行最后还可以包含多个可选字段,字段格式为【标签(两个字符):数据类型(字符、整型、浮点型、字符串等):值】。可选字段的标签非常多,这里只列出几个特殊的标签:
- X0:最优比对的数量(存在多个最最优比对时)。
- X1:次优比对的数量。
- XT:是否唯一比对至此,(Unique/Repeat/N/Multiply)。
- XM:错配(mismatch)的碱基数量。
在NGS分析的过程中,经常要对包含比对信息的SAM/BAM/CRAM格式文件进行基本处理,常见的为bam文件。本文所用工具为SAMtools,常用的SAMtools命令及功能说明如下。
- index:对bam文件建立索引。
- merge:归并多个bam文件为一个。
- cat:连接多个未排序的bam文件。
- sort:将bam文件中的reads按映射位置或名称进行排序。
- depth:计算指定区域的映射深度。
- flagstat:输出bam文件的统计信息
- flags:根据bam文件的位标识字段进行过滤或提取。
- view:对SAM/BAM/CRAM格式的相互转换,还可进行数据切片,保留映射到指定染色体、指定坐标范围内的测序片段。
- tview:测序片段映射情况浏览器,终端界面可视化测序片段的比对信息。