VCF格式说明及常用处理
目前,基因组变异数据通常使用VCF(Variant Call Format)格式以文本形式进行保存,如千人基因组计划或其他机构发布的验证过的变异,此外,许多变异检测工具也通常使用VCF来保存变异检测结果。这里只对VCF格式进行简要的说明(详细官方文档见这里),下图为一示例VCF格式文件。
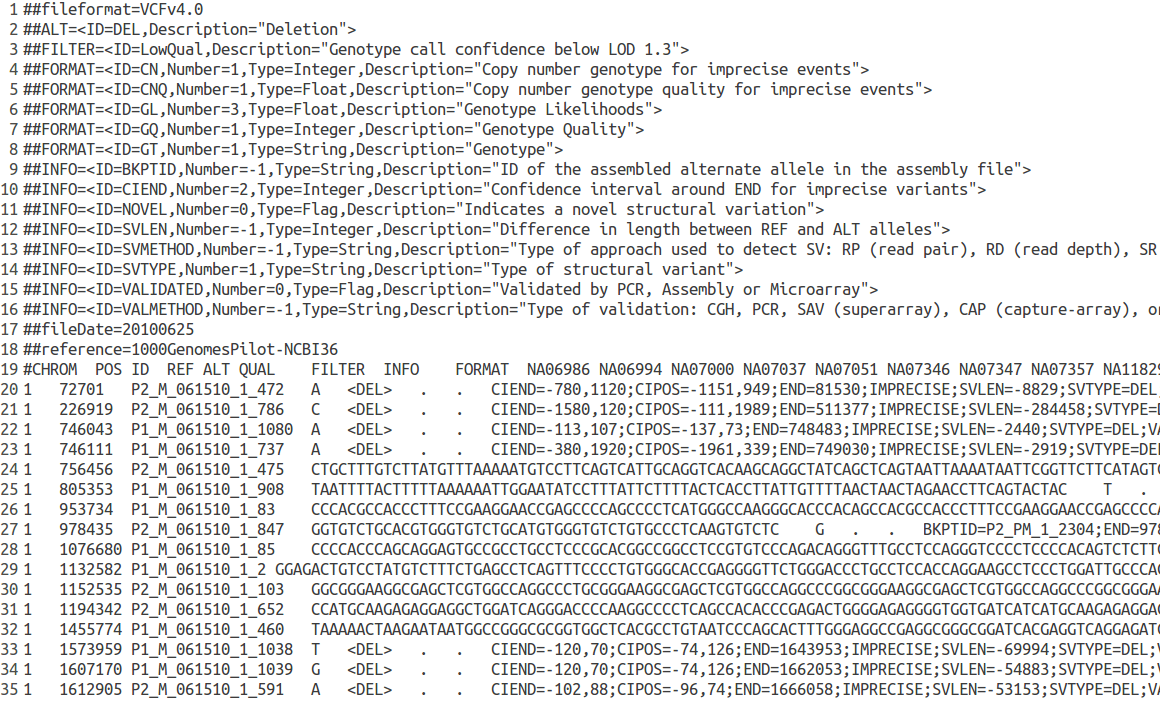
VCF(Variant Call Format)文件格式
同SAM格式一样,VCF格式文件也分为两部分,头部注释和主体两部分。注释信息行以“#”开头,包括该文件的基本信息,如VCF的版本、生成日期、参考基因组等,注释信息的最后一行说明主体部分每个字段的含义,相当于表头的作用。主体部分每行为一个变异,变异的详细信息保存在以“TAB”分割的多个字段中,主要字段包括:
- CHROM:存在变异的染色体号。
- POS:变异在参考序列上的起始坐标。
- ID:变异的ID。
- REF:参考序列的碱基组成。
- ALT:对应变异的碱基组成。
- INFO:附加信息,可包含多个以“;”分割的字段,如END表示变异结束位置,SVLEN表示变异长度,SVMETHOD表示检测方法等。
- 基因型区域:依次列出表头中个体此变异的基因型。
VCF常用处理
通常我们得到VCF文件之后,可能还需要进一步处理,包括分析,过滤和切片等操作。
使用SAMtools的内置工具BCFtools可以完成针对VCF格式文件以及其压缩格式BCF文件的许多操作,包括统计(stat)、过滤(filter)以及merge/cat等操作。类似的工具还有VCFtools。
上述两个工具是功能比较齐全的。如果在千人基因组网站上搜索“slice”会得到如何获取数据文件(BAM/VCF)的子集。其中BAM文件可以使用samtools view -h bamUrl/bamPath chrId:start-end进行切片,配合-f/-F进行过滤筛选操作。VCF文件可以使用tabix进行数据切片,类似命令如tabix -h vcf.gzUrl/vcf.gzPath chrId:start-end | grep ……。当然这些切片操作可能需要事先对文件建立索引,以便于查找染色体位置。除了命令行之外,其基因浏览器上也提供了一个Data Slicer,只是觉得不如命令行好用一些。其实VCF数据文件就是一个文本文件,许多操作可以直接通过grep/awk/sed等文本处理工具实现。
除了简单的过滤切片之外,还有一个处理需求就是截取对应个体存在变异的基因型,用于把Released VCF数据作为Benchmark用于验证变异检测性能,参照VCF数据处理之——截取个体变异的基因型。